Genotype simulation
The GRAB package can be used to simulate genotype data for both unrelated and related subjects. The genotype example data in the directory system.file("extdata", package = "GRAB") is simulated as below line by line (we kept the first 1000 markers to minimize the package size).
Quick Start-up Guide
set.seed(12345)
OutList = GRAB.SimuGMat(nSub = 500, # 500 unrelated subjects
nFam = 50, # 50 families
FamMode = "10-members", # each family includes 10 members
nSNP = 10000, # 10000 SNPs
MaxMAF = 0.5, MinMAF = 0.05) # MAFs follow a uniform distribuiton U(0.05, 0.5)
The function GRAB.SimuGMat returns an R list OutList including two elements of GenoMat and markerInfo as below.
summary(OutList)
# Length Class Mode
# GenoMat 10000000 -none- numeric
# markerInfo 2 data.table list
markerInfocontains two columns:SNPandMAF
markerInfo = OutList$markerInfo
markerInfo
# SNP MAF
# 1: SNP_1 0.3744068
# 2: SNP_2 0.4440979
# 3: SNP_3 0.3924420
# 4: SNP_4 0.4487561
# 5: SNP_5 0.2554164
# ---
# 9996: SNP_9996 0.3270282
# 9997: SNP_9997 0.2866840
# 9998: SNP_9998 0.0601416
# 9999: SNP_9999 0.2161107
# 10000: SNP_10000 0.1632723
GenoMatis a matrix, each row is for one subject and each column is for one SNP.
GenoMat = OutList$GenoMat
dim(GenoMat)
# [1] 1000 10000 # genotype matrix includes 1000 subjects and 10000 SNPs
class(GenoMat)
# [1] "matrix" "array"
GenoMat[c(1:5,996:1000),1:10] # Subjects `f1_1` - `f1-10` are from family 1; `Subj-1` - `Subj-500` are unrelated subjects
# SNP_1 SNP_2 SNP_3 SNP_4 SNP_5 SNP_6 SNP_7 SNP_8 SNP_9 SNP_10
# f1_1 0 1 2 2 1 0 0 0 1 1
# f1_2 1 1 1 0 0 0 0 0 1 1
# f1_3 1 1 0 1 0 0 1 0 1 1
# f1_4 1 1 1 2 1 0 1 2 0 1
# f1_5 0 2 2 1 0 0 0 0 0 1
# Subj-496 1 0 1 0 0 0 1 2 1 1
# Subj-497 1 1 0 0 1 0 1 0 1 0
# Subj-498 0 2 1 1 0 1 1 0 0 0
# Subj-499 2 0 1 1 1 0 0 0 0 1
# Subj-500 1 0 0 2 0 0 0 1 0 2
Note about FamMode
Currently, we support three FamMode including 4-members, 10-members, and 20-members with the family structures as below. If nFam is not specified, then genotype were simulated only for unrelated subjects.
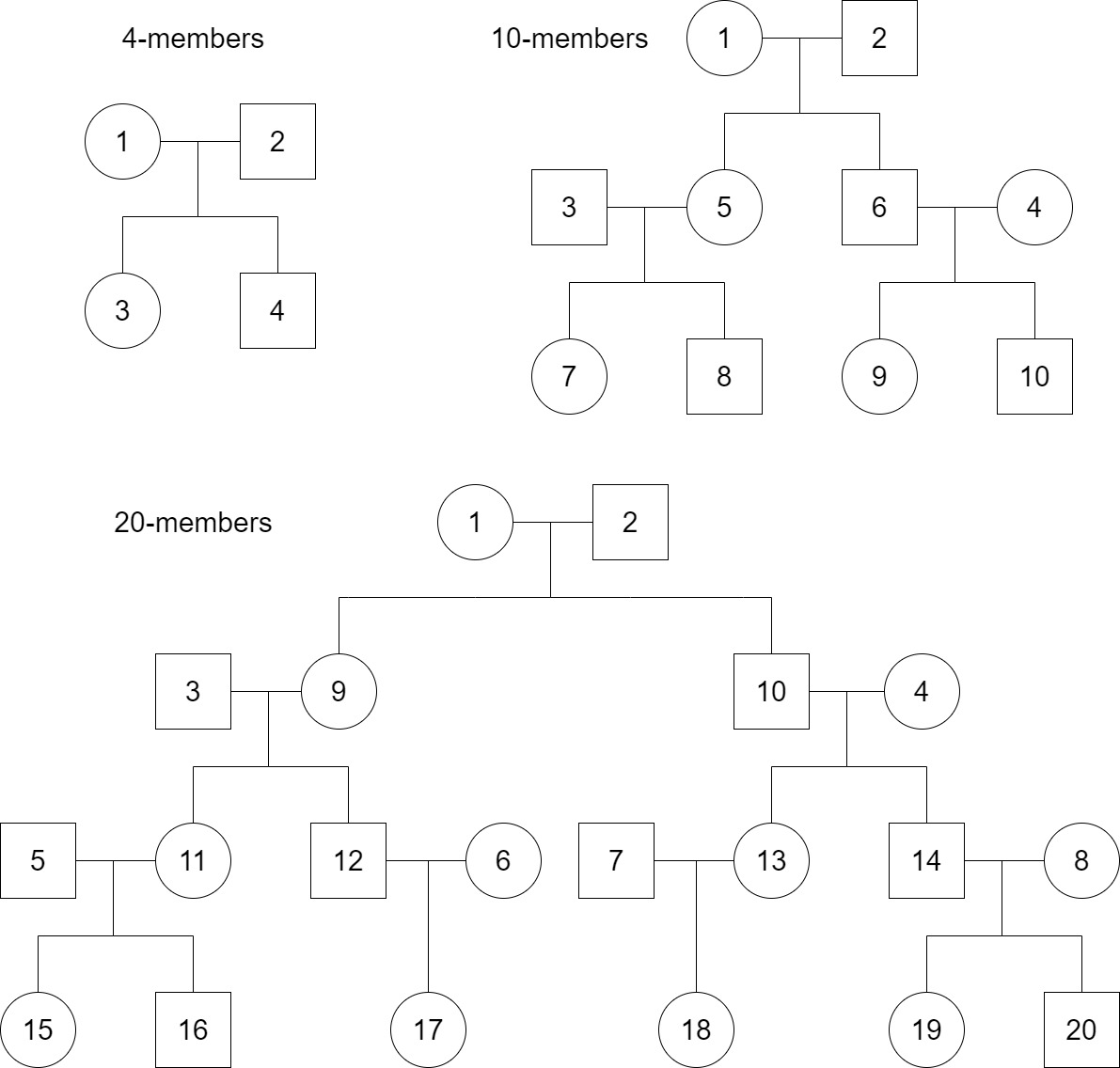
Simulate genotype missing
The below gives an example to simluate genotype missing given a missing rate, in which -9 is to indicate genotype missing, as PLINK does.
MissingRate = 0.05
indexMissing = sample(length(GenoMat), MissingRate * length(GenoMat))
GenoMat[indexMissing] = -9
GenoMat[c(1:5,996:1000),1:10]
# SNP_1 SNP_2 SNP_3 SNP_4 SNP_5 SNP_6 SNP_7 SNP_8 SNP_9 SNP_10
# f1_1 0 1 2 2 1 0 0 0 1 1
# f1_2 1 1 1 0 0 0 0 0 1 1
# f1_3 1 1 0 1 0 -9 1 0 1 1
# f1_4 1 1 1 2 1 0 1 2 0 1
# f1_5 0 2 2 1 -9 0 0 0 0 1
# Subj-496 1 0 1 0 0 -9 1 2 1 1
# Subj-497 1 1 0 0 1 0 1 0 1 0
# Subj-498 0 2 1 1 0 1 1 0 0 0
# Subj-499 2 0 1 1 1 0 0 0 0 1
# Subj-500 1 0 0 2 0 0 0 1 0 2
Make PLINK PED/MAP files using the genotype matrix
Note: this function is not computationally efficient and will be updated later.
extDir = tempdir()
extPrefix = file.path(extDir, "simuPLINK")
GRAB.makePlink(GenoMat, extPrefix)
If you have installed softwares PLINK1.9, PLINK2, and bgenix, then you can use the following commands to generate PLINK binary files and BGEN files.
setwd(extDir)
system("plink --file simuPLINK --make-bed --out simuPLINK")
system("plink --bfile simuPLINK --recode A --out simuRAW")
system("plink2 --bfile simuPLINK --export bgen-1.2 bits=8 ref-first --out simuBGEN") # UK Biobank use 'ref-first'"
system("bgenix -g simuBGEN.bgen -index")
Rare variants simulation (mainly to evaluate set-based approaches): (to be updated: 2022-08-22)
Given arguments of MaxMAF and MinMAF, function GRAB.SimuGMat can simulate
- common variants (MAF > 5%) and
- low frequency variants (1% < MAF < 5%).
For rare variants (MAF < 1%), we suggest using real genotype data from unrelated subjects to simulate family structure while mimicking the LD structure.
Simulate genotype using real data
Function GRAB.SimuGMatFromGenoFile can simulate genotype data using PLINK and BGEN files.
set.seed(123)
nFam = 50
nSub = 500
FamMode = "10-members"
# PLINK data format
PLINKFile = system.file("extdata", "example_n1000_m236.bed", package = "GRAB")
IDsToIncludeFile = system.file("extdata", "example_n1000_m236.IDsToInclude", package = "GRAB")
GenoList = GRAB.SimuGMatFromGenoFile(nFam, nSub, FamMode, PLINKFile,
control = list(IDsToIncludeFile = IDsToIncludeFile))
# Currently, this function does not support BGEN data format
# BGENFile = system.file("extdata", "example_n1000_m236.bgen", package = "GRAB")
# IDsToIncludeFile = system.file("extdata", "example_n1000_m236.IDsToInclude", package = "GRAB")
# GenoList = GRAB.SimuGMatFromGenoFile(nFam, nSub, FamMode, BGENFile,
# control = list(IDsToIncludeFile = IDsToIncludeFile))
Then, we make PLINK files using the genotype data
GenoMat = GenoList$GenoMat
GenoMat[is.na(GenoMat)] = -9
extDir = tempdir()
extPrefix = file.path(extDir, "simuPLINK_RV")
GRAB.makePlink(GenoMat, extPrefix)
setwd(extDir)
system("plink --file simuPLINK_RV --make-bed --out simuPLINK_RV")